DISEASE MODELS
|
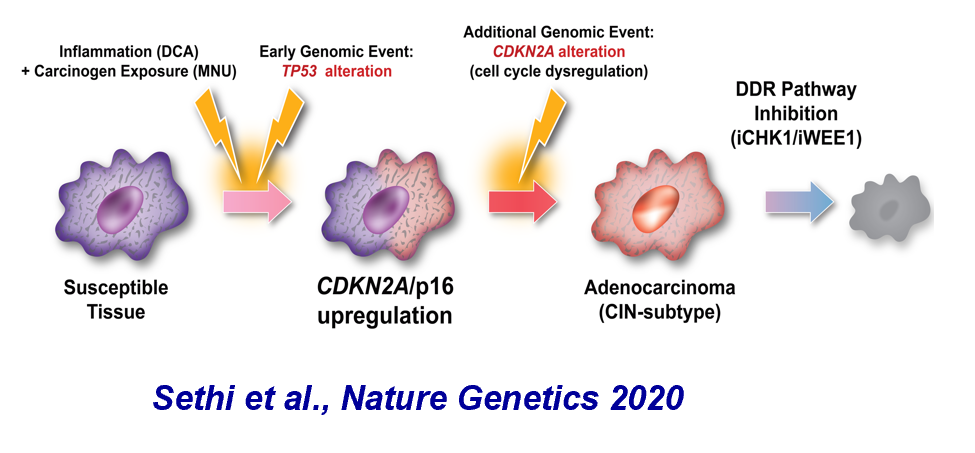
We recently built a new mouse model that combines early genetic alterations with disease-relevant dietary carcinogens to study gastric premalignancy. Using this model, we learned that early TP53 alterations ultimately lead to premalignant lesions that have a selective pressure to alter a cell cycle regulator. Although this second alteration enabled disease progression, it also yielded a therapeutic vulnerability.
|
|
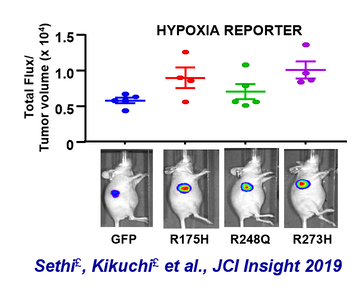
After realizing that hypoxia transcriptional programs may be heightened in p53 mutant gastric and esophageal adenocarcinomas, we designed an in vivo system to measure hypoxia activity in real-time during xenograft tumor growth. Using this system, we were able to identify a new dependency in these deadly cancers. |
|
|
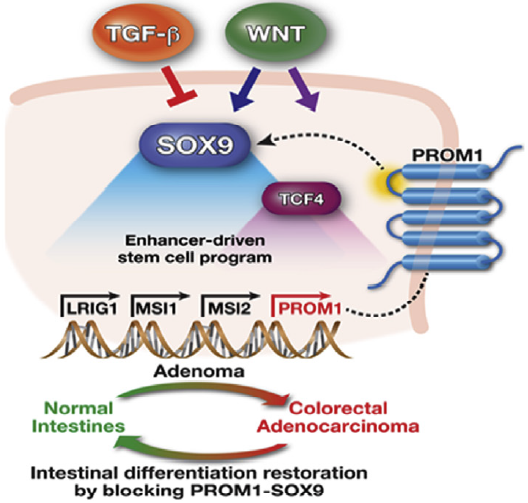
Using a combination of patient specimen histopathology, human cancer cell lines, and mouse organoid models, we demonstrate that SOX9 promotes colorectal cancer by restricting differentiation. By engaging genome-wide enhancers, SOX9 directly activates a stem-cell like program. SOX9 induces stem cell factor PROM1 through an intronic enhancer. PROM1 expression is sufficient to block differentiation in colorectal cancer via its first intracellular domain. Disrupting either SOX9 or PROM1 reduces tumor growth by inducing differentiation. These studies implicate SOX9-mediated differentiation impairment as an early, important step in colorectal cancer development and inspire pro-differentiation therapeutic approaches for the treatment of this deadly disease.
Liang et al., Gastroenterology, 2022 |
|
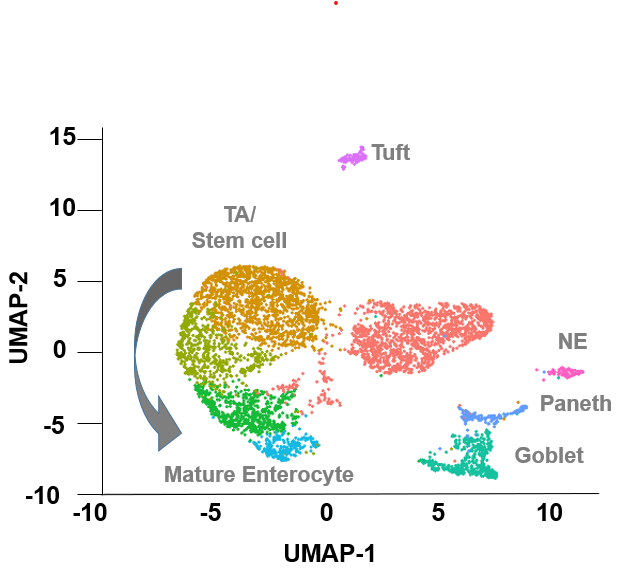
We have designed a genetically engineered mouse model to characterize the functional and clinical significance of SOX9 alterations in colorectal cancer. Phenotypic evaluation, histopathological analyses, and molecular studies (e.g. single-cell RNA-sequencing) of this model will enable a thorough understanding of the genome stable subtype of colorectal cancer.
|
|